Hai...... Good morning to all....
Yesterday I forgot to add Mass balance concept to forced degradation study.
So today my concept is.......
Yesterday I forgot to add Mass balance concept to forced degradation study.
So today my concept is.......
" Mass balance calculation during Forced degradation study"
There is no proper guidance from any regulatory authorities for mass balance calculation during forced degradation study.
The mass balance concept is based on the "law of conversion of mass" which states "Mass can neither created nor destroyed"
Mass balance means...?
I will give a simple example with a small story. Understand carefully.
" A college is having exctaly 100 students. Oneday, campus interview was conducted in that college. Out of 100 students, 11 students were selected in campus interview, and 3 students were kept on hold for selection.
Now, Total no. of students = 100
No. of students not selected in interview = 86
No of students selected = 11
No. of students kept on hold for selection = 3.
Total No. of students = (No. of students not selected + No. of students selected + No. of students on hold)
Before coming to the main point, Now lets compare the story with with chemistry.
College = Stability indicating Method
Campus interview = Forced degradation study
Total No. of Students = amount of mass (analyte) taken for degradation
No. of students not selected in interview = amount of mass (analyte) remained after degradation
No of students selected = amount of known degradants observed.
No. of students kept on hold for selection = amount of unknown degradants observed.
Now mass balance means,
Total mass = (amount of mass remained + amount of known degradants + amount of unknown degradants)
Why Mass balance required...?
While developing stability indicating method for degradants ( generally called as RS method), we have to ensure whether the method is exactly quantifying all possible degradants or not. Hence, Mass balance is useful element for that.
How Mass balance calculated..?
As I already explained the mass balance equation above, it is
Total mass = (amount of mass remained + amount of known degradants + amount of unknown degradants)
So, here I will convert the equation for better understanding. After each degradation study,
Total % of drug = % of drug remained + % of known degradants + % of unknown degradants
Important : Don't ran away without knowing this points...
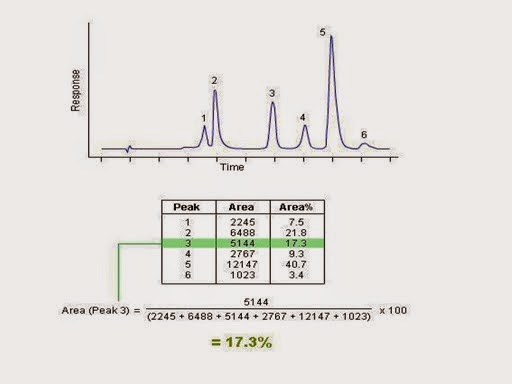
1) Relative response factors plays an important role to calculate exact mass balance.
2) Molecular weight of Drug and molecular weight of degradant formed plays an important role to calculate exact mass balance.
Importance of Relative Response Factor(RRF):
Suppose for example, if the RRF of an impurity (RRF should be established while developing analytical method) is 0.6.
and the if amount of degradant formed as per area calculation is 3.5, . now the exact amount of impurity formed will be
= 3.5 * 1/0 .6 = 5.83%
Importance of Molecular weight(MW):
1) Suppose for example, MW of Main drug is 151, and MW of degradant formed is 195. If the % of degradant formed as per % area is 3.5, now the exact amount of impurity formed is
3.5* 151/195 = 2.71%
2) Suppose for example, MW of Main drug is 151, and MW of degradant formed is 125. If the % of degradant formed as per % area is 3.5, now the exact amount of impurity formed is
3.5* 151/125 = 4.23 %
What is limit for mass balance.?
The ultimate goal is that the analytical method should achieve 100% mass balance. however, the acceptable limit for mass balance will be not less than 95%.
What to do if mass balance not acheived.?
Some times 100% mass balance may not be achieved because of
1) The degradant formed may not be eluted in the developed method.(extend runtime or modify method conditions)
2) The RRF may be zero at method wavelength.(select appropriate wavelength or develop separate method)
3) Degradant may be UV inactive.( Identify the impurity by different techniques like RI, ELSD, LC-MS etc..)
Don't worry if the mass balance is not achieved, proper justification should be given for not achieving mass balance. as I shown in the brackets for each above points.
That's it for today........... have a nice day.........
V.Suresh
The mass balance concept is based on the "law of conversion of mass" which states "Mass can neither created nor destroyed"
Mass balance means...?
I will give a simple example with a small story. Understand carefully.
" A college is having exctaly 100 students. Oneday, campus interview was conducted in that college. Out of 100 students, 11 students were selected in campus interview, and 3 students were kept on hold for selection.
Now, Total no. of students = 100
No. of students not selected in interview = 86
No of students selected = 11
No. of students kept on hold for selection = 3.
Total No. of students = (No. of students not selected + No. of students selected + No. of students on hold)
Before coming to the main point, Now lets compare the story with with chemistry.
College = Stability indicating Method
Campus interview = Forced degradation study
Total No. of Students = amount of mass (analyte) taken for degradation
No. of students not selected in interview = amount of mass (analyte) remained after degradation
No of students selected = amount of known degradants observed.
No. of students kept on hold for selection = amount of unknown degradants observed.
Now mass balance means,
Total mass = (amount of mass remained + amount of known degradants + amount of unknown degradants)
Why Mass balance required...?
While developing stability indicating method for degradants ( generally called as RS method), we have to ensure whether the method is exactly quantifying all possible degradants or not. Hence, Mass balance is useful element for that.
How Mass balance calculated..?
As I already explained the mass balance equation above, it is
Total mass = (amount of mass remained + amount of known degradants + amount of unknown degradants)
So, here I will convert the equation for better understanding. After each degradation study,
Total % of drug = % of drug remained + % of known degradants + % of unknown degradants
Important : Don't ran away without knowing this points...
1) Relative response factors plays an important role to calculate exact mass balance.
2) Molecular weight of Drug and molecular weight of degradant formed plays an important role to calculate exact mass balance.
Importance of Relative Response Factor(RRF):
Suppose for example, if the RRF of an impurity (RRF should be established while developing analytical method) is 0.6.
and the if amount of degradant formed as per area calculation is 3.5, . now the exact amount of impurity formed will be
= 3.5 * 1/0 .6 = 5.83%
Importance of Molecular weight(MW):
1) Suppose for example, MW of Main drug is 151, and MW of degradant formed is 195. If the % of degradant formed as per % area is 3.5, now the exact amount of impurity formed is
3.5* 151/195 = 2.71%
2) Suppose for example, MW of Main drug is 151, and MW of degradant formed is 125. If the % of degradant formed as per % area is 3.5, now the exact amount of impurity formed is
3.5* 151/125 = 4.23 %
What is limit for mass balance.?
The ultimate goal is that the analytical method should achieve 100% mass balance. however, the acceptable limit for mass balance will be not less than 95%.
What to do if mass balance not acheived.?
Some times 100% mass balance may not be achieved because of
1) The degradant formed may not be eluted in the developed method.(extend runtime or modify method conditions)
2) The RRF may be zero at method wavelength.(select appropriate wavelength or develop separate method)
3) Degradant may be UV inactive.( Identify the impurity by different techniques like RI, ELSD, LC-MS etc..)
Don't worry if the mass balance is not achieved, proper justification should be given for not achieving mass balance. as I shown in the brackets for each above points.
That's it for today........... have a nice day.........
V.Suresh